亚砜是一类重要的有机硫化合物,常用作合成制药、农药、溶剂和香料的反应中间体。硫化物的直接选择性氧化是合成亚砜最有效的方法之一。然而,传统氧化方法通常依赖于化学氧化剂(H₂O₂、t-BuOOH、HNO₃、NaOCl)、无机氧化物(MnO₂或Fe(NO₃)₃)和氧气(O₂)等强氧化物,伴随着包括低原子经济性、高碳足迹、爆炸风险、严重的过度氧化以及底物范围有限等一系列挑战。以水为绿色氧源,利用可再生电能驱动的电化学选择性有机氧化,为传统的硫化物到亚砜制备方法提供了一个有力的替代方案,以满足现代清洁能源技术和绿色化学合成对可持续性、风险管理和能源效率的需求。根据热力学分析结果(图1a),水氧化(活化)过程是一个自由能升高的过程,以水在RuO₂(110)上的反应生成氧气为例,该反应涉及多种中间体的转化(H₂O→*OH→*O→*OOH→O₂),其中*OH的氧化能力不足以氧化硫化物,而*OOH和O₂物种存在过度氧化问题和OER副反应,难以实现高选择性和法拉第效率,因此*O物种是一个潜在高效硫醚氧化物种。
在电化学氧化方法中,基于分子基氧化还原媒介体生成的氧活性物种可通过均相催化过程实现硫醚的氧化。然而,均相过程中存在的分离回收等额外成本,严重限制了它们的进一步应用(图1b,左图)。分子基催化媒介由于其稳定性和反应性方面的内在限制,存在工作电化学电位窗口较窄的问题。当需要高电流密度和流动池配置以实现可扩展的化员工产时,这些问题尤为突出。直接电催化氧化策略用于选择性氧化甲基苯基硫化物可以有效避免上述问题。其中,水分子的表面活化是十分关键的步骤,该过程提供“绿色氧”原子插入反应物分子中。但是,与均相催化相比,非均相的直接电有机氧化策略通常在非水有机反应介质中遭受缓慢的单电子转移(SET)和氧(水活化)动力学,导致严重的过电位,目前尚未实现所需的反应活性和选择性(图1b,右图)。这些动力学障碍导致了许多问题,包括低电流密度、低选择性和低法拉第效率,阻碍了电有机氧化的广泛应用,尤其是在电催化硫化物到亚砜的转化中(图1c,左图)。
为解决低水浓度环境中的水氧化动力学缓慢的问题,太阳成集团丁梦宁团队和香港城市大学黄勃龙团队、中国科学技术大学林岳团队合作,设计合成了一种通过增强无序的Ru−O6八面体表面氧动力学实现硫醚高效的电氧化为亚砜,实现了适用于多种亚砜的高效高选择性绿色电催化氧化通用方法。该工作发表在JACS期刊上,题为《Disordered Ru–O6 Octahedrons for Efficient and Selective Electro-oxidation of Sulfide to Sulfoxide via Boosted Surface Oxygen Kinetics》。太阳集团tcy87222022级直博生冉攀等为该工作的共同第一作者。
该工作设计合成了一种介孔非晶态RuO₂纳米片,由特征性的无序连接的Ru-O6八面体组成(命名为a-RuO₂-dO)。研究表明,无序排列的Ru-O6八面体结构大大促进了水分子在低阳极电位下的解离,增加了活性氧物种(*O)的表面覆盖率,从而显著加速了水氧化动力学和随后的硫化物选择性氧化(图1c,右图)。以甲基苯基硫化物(MPS)为模型底物,a-RuO₂-dO在相对较低的氧化电位(有机介质中1.25 V vs. Ag/AgCl)下表现出卓越的性能,在温和条件下,实现了甲基苯基亚砜(MPSO)接近100%的产率(99%)、选择性(98%)和法拉第效率(95%)。此外,在高电流密度(>100 mA/cm²)下广泛的底物范围和克级生产进一步证明了这种电催化合成方法的实用价值。机理和理论研究表明,无序排列的Ru-O八面体单元在增强费米能级附近的成键轨道分布和电子耦合方面起着关键作用,从而提高了表面水氧化的关键决速基元步骤(*OH→*O)和随后的硫化物氧化步骤(*O+MPS→MPSO)的反应动力学,整个催化过程的微反应动力学模型遵循吸附物演化机制介导的Eley-Rideal反应(AEM-ER)路径。这种独特结构促进了更高效的电子转移过程,并降低了关键反应步骤的活化能垒,从而在MPS氧化中表现出优异的催化性能。这些结果突显了原子(簇)无序化在精准催化活性调控和优化过程中的关键作用,使a-RuO₂-dO成为选择性硫化物氧化的高效催化剂。
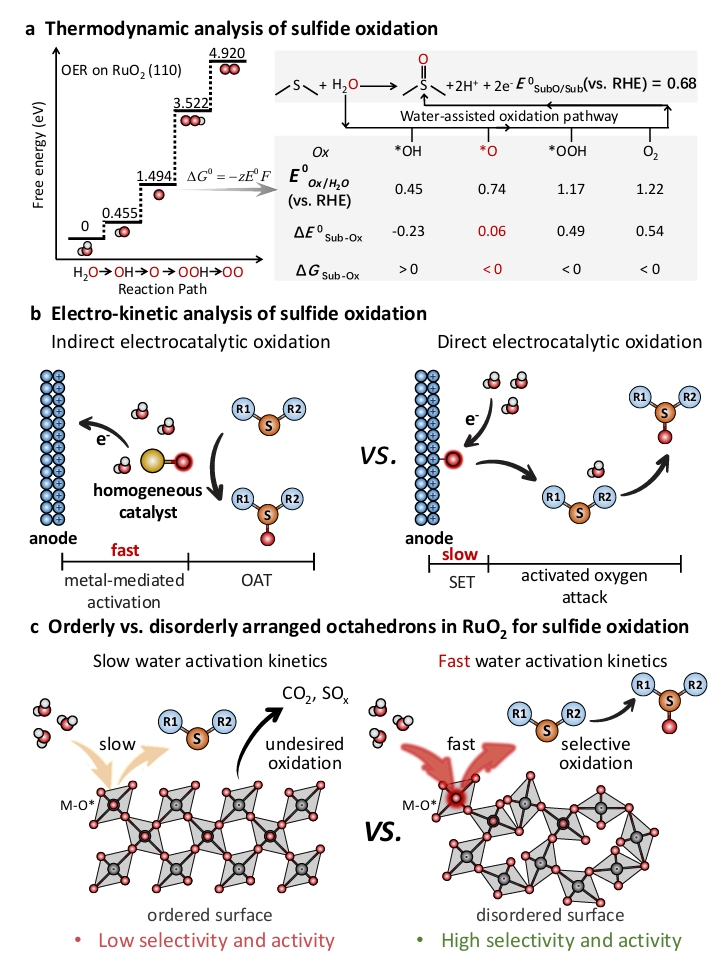
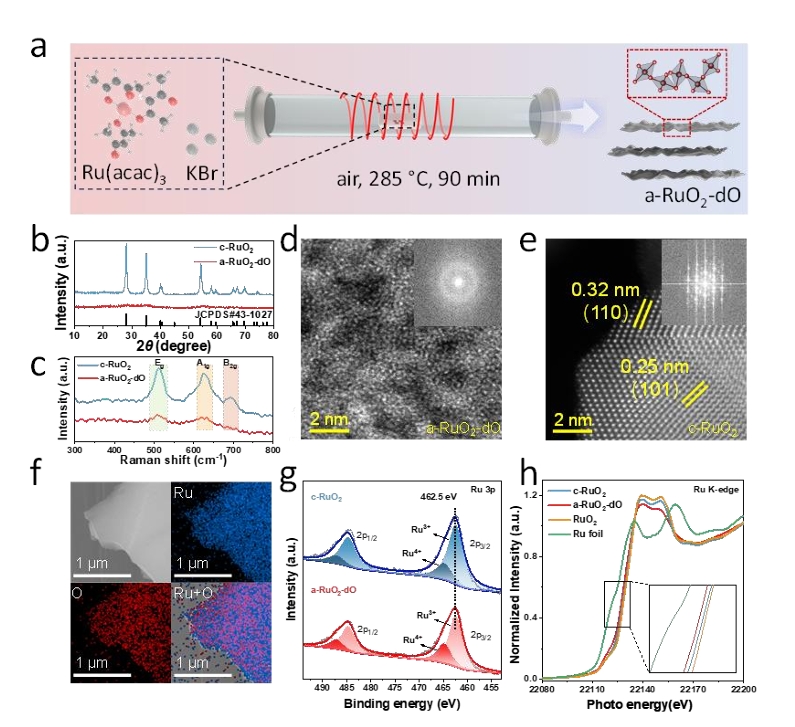
图2. a-RuO2-dO的制备与表征。(a)a-RuO2-dO的合成过程示意图。(b)c-RuO2和a-RuO2-dO的XRD图谱。(c)c-RuO2和a-RuO2-dO的拉曼光谱。(d)a-RuO2-dO的球差校正HAADF-STEM图像(d)和c-RuO2(e),插图显示相应的选区电子衍射图。(f)a-RuO2-dO的SEM图像和元素映射结果。(g)c-RuO2(上)和a-RuO2-dO(下)的Ru 3p XPS分析。(h)a-RuO2-dO、c-RuO2和标准样品的Ru K边XANES光谱。
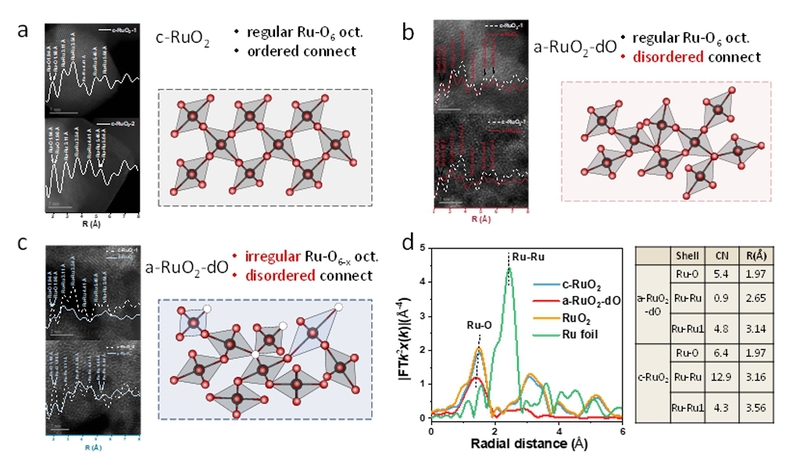
图3. c-RuO2和a-RuO2-dO的结构分析。(a)晶态氧化钌的相位对比校正高分辨透射电镜(HRTEM)的径向分布函数(RDF)分析,右侧为相应的Ru-O结构示意图。(b)非晶态RuO2的HRTEM的RDF分析,右侧为无序连接的规则Ru-O6八面体的示意图。(c)非晶态RuO2的HRTEM的RDF分析,右侧为无序连接的不规则Ru-O6八面体的示意图。(d)a-RuO2-dO和c-RuO2的扩展X射线吸收精细结构(EXAFS)光谱,表格展示了不同样品在Ru K边的EXAFS拟合参数。
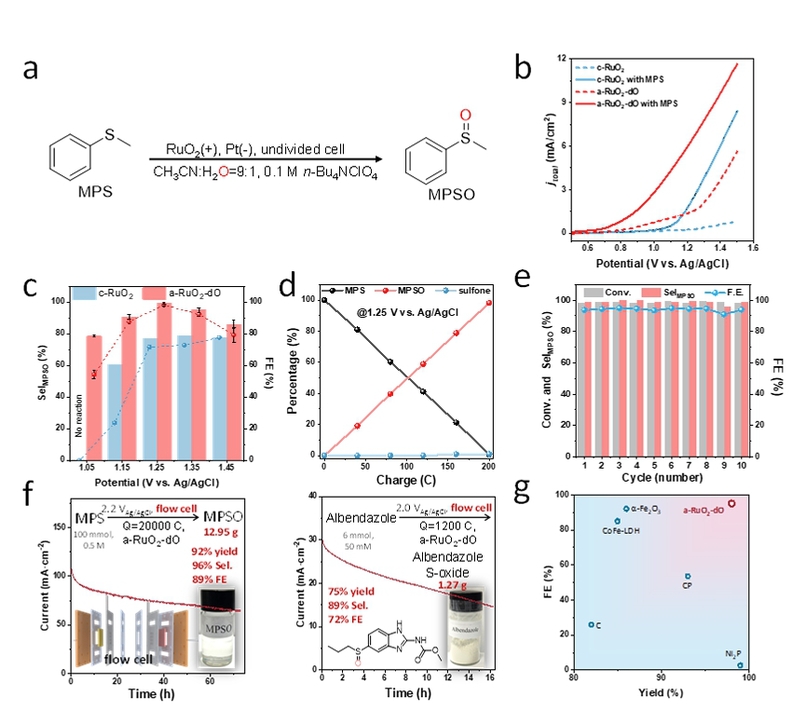
图4. 硫化物选择性氧化为亚砜的电催化性能。(a)模型反应条件,RuO2代表c-RuO2和a-RuO2-dO催化剂。(b)a-RuO2-dO和c-RuO2的线性扫描伏安法(LSV)。(c)在不同施加电位下使用a-RuO2-dO和c-RuO2进行MPS到MPSO转化的法拉第效率(FE)和选择性。(d)MPS、MPSO和砜的百分比与消耗电荷之间的关系。在完成一个标准反应循环后,更换电解液,并使用相同催化剂开始新的电解循环。每个循环对应约25小时的电解。(f)克级反应(g)与最先进的非均相电催化硫化物氧化反应的产率和法拉第效率的比较。(c)中的误差条表示多次反应的标准偏差(n=3)。
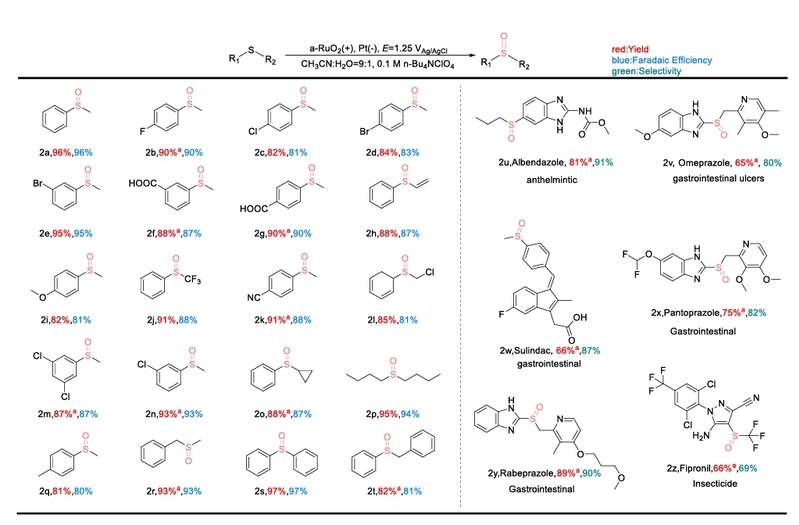
图5. 底物拓展
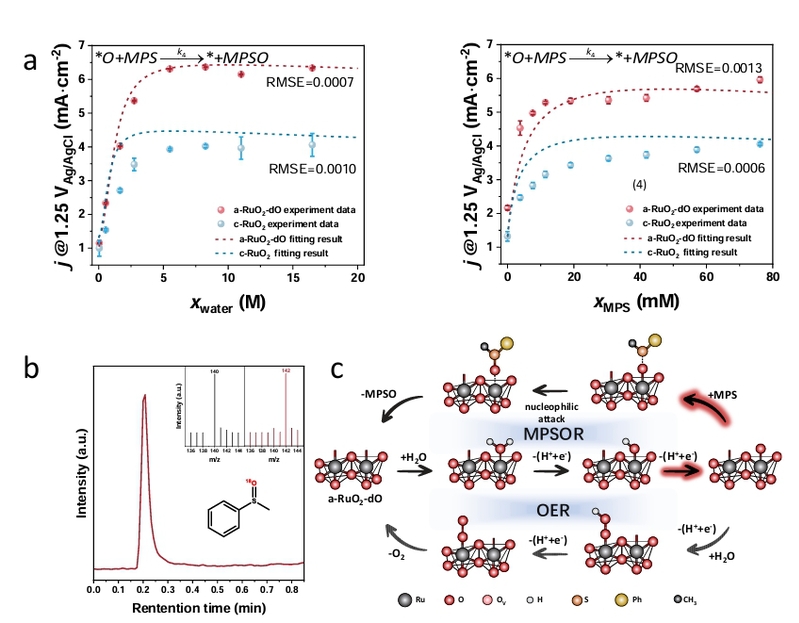
图5. a-RuO2-dO的机理研究。(a)a-RuO2-dO和c-RuO2催化剂的微动力学分析,左图显示水依赖的动力学,右图显示MPS依赖的动力学。数据点表示实验部分电流密度(相当于高法拉第效率下的反应速率)对水/底物浓度的依赖关系,虚线曲线表示动力学数据的数学拟合。底部显示的反应速率方程是从微动力学模型的构建中得出的。(a)中的误差条表示多次反应的标准偏差(n=3)。(b)在CH3CN-18O标记的水和CH3CN-水中的MPS电氧化的MPSO质谱。(c)在a-RuO2-dO上直接硫化物到亚砜氧化(MPSOR)和氧析出反应(OER)的吸附物演化机制(AEM)的提出。
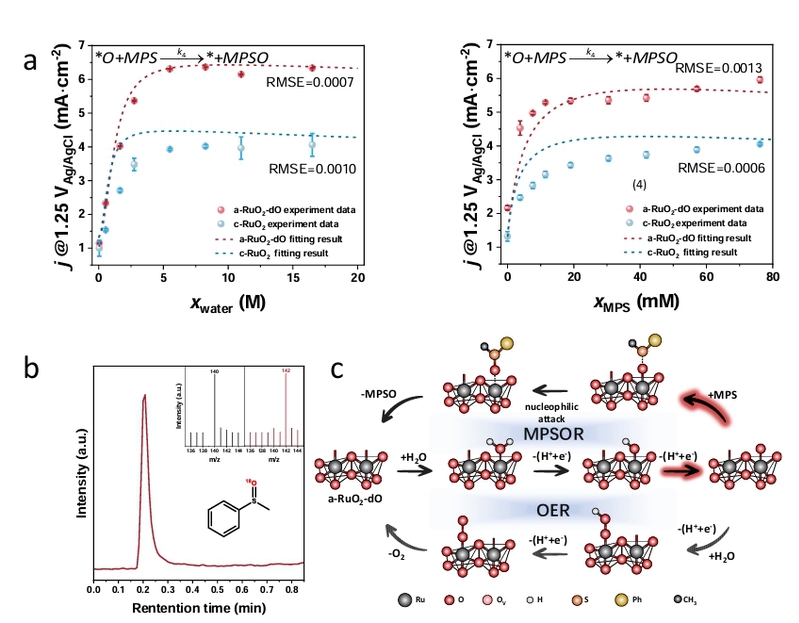
图6. 在(a)晶态RuO₂和(b)非晶态RuO₂上费米能级附近成键和反键轨道的电子分布。蓝色球体 = Ru,红色球体 = O。蓝色等值面 = 成键轨道,绿色等值面 = 反键轨道。(c)非晶态RuO₂的投影态密度(PDOS)。蓝色球体 = Ru,红色球体 = O。(d)非晶态RuO₂中Ru-4d和(e)O-s,p的位点依赖PDOS。(f)在非晶态RuO₂中AEM和LOM路径下关键中间体的PDOS。(g)MPS和H₂O的吸附能比较。(h)通过AEM路径在晶态RuO₂(红色)和非晶态RuO₂-dO(蓝色)上MPS氧化的反应能图。
综上所述,本工作突显了原子无序在克服催化剂优化过程中常见动力学限制方面的独特和有效作用,从而实现了非水介质中理想的直接选择性电氧化有机物。该成果为非水体系中有机物的高效选择性电氧化提供了重要的微反应动力学设计原理。该研究工作得到了国家自然科学基金、江苏省自然科学基金、中央高校基础研究基金、中国科公司青年创新促进会、国家重点研发计划、前沿科学重点研究计划和介观化学教育部重点实验室的支持。